第五节 某些肝病的生化机制


一、乙醇在肝内的代谢及乙醇性肝损伤
乙醇对于人体来说是一种异物,在肠道内由于细菌发酵所产生的乙醇仅以微量存在,因而对机体影响不大。我们所讲的乙醇代谢主要是指通过饮酒而摄入体内的外源性乙醇在体内的代谢。乙醇在胃及小肠上部迅速被吸收(胃30%,小肠上部70%),被摄取的乙醇90%-98%在肝内被代谢,剩下的2%-10%随尿及呼出气而被排泄。人的乙醇代谢率从其在血中浓度的消失率来看为100-200mg(kg·h)。因而在健康成人为每小时10g左右,一日代谢量约为240g 。
乙醇的饮用量与肝硬化的发病率之间有密切关系,饮酒量越高的国家,其肝硬化死亡率也越高。例如饮酒量每人在10升/年以下的瑞典、英国、丹麦等国家,肝硬化的死亡率在10/10万以下;而饮酒量最高的法国人均饮酒量27升/年左右,其肝硬化死亡率则为30/10万以上。由于长期饮酒容易形成乙醇性脂肪肝、乙醇性肝炎、肝硬化,甚至在孕妇还可造成胎儿性乙醇综合征,影响胎儿的发育成长。因此,了解乙醇在人体的正常代谢及乙醇对有机体的影响,在含酒精饮品日益泛滥的今天无疑具有重要的生理意义、临床意义和社会意义。
㈠乙醇在体内的代谢
乙醇在体内的代谢具有下述特征:①乙醇在作为药物(异物)的同时,每克能释放7Kcal(1cal=4.2J)的热能;②被摄取的乙醇的大部分(90%-98%)被代谢,由肾和肺排泄的仅占一小部位;③乙醇的大部分在肝脏内被氧化;④乙醇及其代谢产物不能在体内储存;⑤并不存在调节乙醇氧化速度的特殊的反馈机制。
乙醇的代谢途径包括乙醇脱氢酶(alcohol dehydrogenase,ADH)催化的乙醇氧化体系(即ADH乙醇氧化体系)与微粒体乙醇氧化体系(microsomal ethanol oxidizing system,MEOS),另外还有NADPH氧化酶-过氧化氢酶体系以及黄嘌呤氧化酶-过氧化氢酶体系(表10-14)。这些体系中以ADH乙醇氧化体系与微粒体乙醇氧化体系最为重要。
表10-14 肝脏中的乙醇代谢体系

⒈ADH乙醇氧化体系被摄取至肝内的乙醇大部分被肝细胞液中的乙醇脱氢酶催化脱氢而生成乙醛,乙醛进一步在乙醛脱氢酶催化下脱氢而生成乙酸,后者又形成乙酰辅酶A而进入三羧酶循环,最后生成二氧化碳和水,并释放能量生成ATP。
乙醛脱氢酶可分为两型,Ⅰ型为NAD依赖性的低Km酶,全分布在线粒体内;Ⅱ型为高Km酶,分布在线粒体及微粒体中。乙醇氧化产生的乙醛大部分在线粒体内被NAD依赖性的低Km酶的代谢系统所氧化。
⒉微粒体乙醇氧化体系(MEOS)乙醇的代谢与肝细胞微粒体的功能有很大关系,从形态学上观察到:长期饮酒的人及实验动物的肝细胞滑面内质网显著增加,表明乙醇也可能在微粒体被代谢。ADH乙醇氧化体系与微粒体乙醇氧化体系的组成和性质不同(表10-15)。
表10-15 乙醇脱氢酶(ADH)体系与微粒体乙醇氧化体系(MEOS)的比较
| ADH体系 | MEOS | |
| 细胞内的区域性分布 | 肝细胞胞液、线粒体内 | 肝细胞微粒体 |
| 最适pH | 10.8 | 7.2-7.4 |
| 辅酶 | NAD+ | NADPH |
| 米氏常数(Km) | 2mmol/L | 8.6mmol/L |
| 吡唑的抑制程度 | 抑制99% | 抑制3% |
| (4.4mM/kg体重) | ||
| 投与乙醇后引起的活性变化 | 不变 | 增加 |
| 与乙醇氧化相伴的能量变化 | 与氧化磷酸化相偶连产生氢,释能 | 需NADPH和O2,耗能 |
| 在乙醇代谢中所占的比率 | 75%-80% | 20%-25% |
1970年Lieber与De Carli用大鼠及人肝微粒体对MEOS的特征进行探讨,发现MEOS的反应有以下特征:①最适pH是在生理范围(pH6.8-7.4)内;②对底物(乙醇)的Km为8.2mmol/L;③需要NADPH和O2作为辅助因子,如用其它吡啶核苷酸作为电子供体时则不能反应;④对CO气体较敏感;⑤对过氧化氢酶、ADH两种酶的抑制剂较有耐受性;⑥长期饮酒对MEOS系统可明显产生诱导。
㈡乙醇代谢对机体的影响
乙醇代谢亢进所并发的各种代谢异常可能是由于NADH/NAD+比值的上升,因此先加以叙述。
⒈NADH/NAD+比值的上升乙醇在体内的代谢第一阶段是由乙醇生成乙醛,第二阶段是由乙醛氧化成乙酸。催化这两步反应的乙醇脱氢酶、乙醛脱氢酶的辅酶都是NAD+。因此,在乙醇氧化的过程中NAD+被还原为NADH。通过乙醇代谢产生的过剩的NADH制成NADH/NAD+比值的上升或NAD+/NADH比值的降低。例如给予2.4g/kg体重乙醇处理时,在给乙醇两小时后,NAD+/NADH比值从4.1降至1.5。NADH/NAD+比值的上升(或NAD+/NADH比值的下降)使得肝脏中乳酸的利用降低。另一方面,丙酮酸被增多的NADH还原成乳酸,容易导致乳酸性酸中毒。又可由于酸中毒引起肾排泄尿酸的障碍,导致发生高尿酸血症。
⒉乙醛对机体的影响在乙醇代谢对机体造成影响的因素中,不能忽视乙醇在肝内代谢的中间产物-乙醛的作用。乙醛的化学性质活泼,具有强烈的药理作用。动物实验表明:摄取乙醇四周的大鼠对乙醛的氧化率明显低于对照组,这可能是由于乙醇引起线粒体的功能障碍所致。长期饮酒所引起的肝线粒体的功能不全可能是因为乙醛的毒性所致。乙醛可引起线粒体的功能障碍,使线粒体的呼吸功能、脂肪酸氧化能力受到损伤。在慢性饮酒者可能形成一种恶性循环:持续饮酒可引起肝内产生较多的乙醇,高浓度的乙醛引起肝细胞线粒体的损伤,于是线粒体的乙醛代谢率降低,导致肝内乙醛浓度进一步上升,线粒体功能进一步低下。
乙醛除了对脑内胺代谢、线粒体呼吸功能、心肌蛋白质合成能力等有抑制作用外,还具有下述药理作用或毒性作用:①乙醛具有使内源性儿茶酚胺释放的刺激交感神经样作用,这可能是引起乙醇性心肌病的一个原因;②乙醛可能是四乙秋兰姆化二硫(戒酒硫,disulfiram)引起的中毒作用的原因;③乙醛与儿茶酚胺缩合成了与吗啡生物碱的前身物质结构非常相似的四氢异喹啉,这一物质是酒瘾发病的原因;④乙醛使5-羟色胺代谢发生障碍,结果产生具有幻觉作用的四氢-β-咔啉,可引起酒后的种种精神障碍;⑤乙醛是乙醇引起的戒断症状发病的一个原因;⑥乙醛对肝和脑的辅酶A活性具有相同抑制作用;⑦乙醛是造成慢性饮酒者维生素B6缺乏症的重要成因;⑧乙醛能抑制脑内Na+、K+-ATP酶。L-抗坏血酸、L-半胱氨酸等可防止乙醛对ATP酶的抑制作用。
⒊乙醇对血糖、氨基酸代谢、水电解质平衡、维生素D代谢及药物代谢的影响饮酒后血糖有降低倾向,健康人在较长时间(48-72小时)饥饿状态下给予乙醇能发生显著的低血糖。这可能是由于NADH的增高导致丙酮酸氧化性-脱羧、脂肪酸氧化、三羧酸循环等过程发生障碍,饥饿状态下摄取乙醇时糖异生过程亦发生障碍,加上摄取乙醇时进食不足,造成肝糖原储备降低,这皆是导致乙醇性低血糖的原因。
喂乙醇动物有负氮平衡倾向,乙醇性肝损伤时有蛋白质代谢障碍,慢性投予乙醇,动物血中γ-谷氨酰转肽酶活性增高。嗜酒者血清谷氨酸脱氢酶(线粒体酶)活性显著上升,表明乙醇性肝损伤时,肝小叶中心部肝细胞线粒体发生损伤,因为谷氨酸脱氢酶主要分布于肝小叶中心部位。
饮酒后利尿作用增强,伴脱水症状,引起口渴。乙醇利尿现象是垂体抗利尿激素分泌的抑制所致。血清中各种电解质因脱水而浓缩,饮酒后还引起血液pH降低、重碳酸盐减少,有酸中毒倾向,这主要是由于乙醇代谢时引起的乳酸、乙酸、酮体等增加所致。
乙醇性肝损伤(如乙醇性脂肪肝、乙醇性肝炎、乙醇性肝硬化)时均可观察到血清中25-OH-维生素D的减低,其降低的原因主要是维生素摄取不足、乙醇作用于肠道引起的吸收障碍、肝中25-羟化作用的降低及分泌的降低等。
当给急性乙醇中毒患者镇静剂和安眠药时可见到敏感的反应,有时会产生意想不到的重度中毒症状。另一方面慢性饮酒者可对镇静剂呈抗药性,麻醉剂的效果亦较难出现。在肝内代谢、对肝有毒性的CCl4,如果给予慢性摄取乙醇的动物,则会显示更强的毒性。解热镇痛剂乙酰氨基酚在对照组不发生肝损坏的剂量,对慢性乙醇投入组则可导致肝细胞坏死。乙醇与药物同在微粒体中被代谢,当乙醇与药物同时进入人体时,即可能发生对于其共同氧化系统细胞色素P450的底物的竞争现象。另外大量乙醇可引起药物代谢酶活性的抑制,这样,乙醇与药物两者的代谢是相互抑制的。于是会引起用药时发生意外的严重的中毒症状。因此,大量应用镇静剂、安眠药时必须注意患者有否饮酒嗜好。
(三)乙醇性肝损伤与胎儿性乙醇综合征
本段着重叙述几种重要的乙醇性肝损伤:乙醇性脂肪肝、乙醇性肝炎、乙醇性肝硬化以及妊娠时由于孕妇饮酒而造成胎儿异常的胎儿性乙醇综合征(fetal alcohol syndrome,FAS)。
⒈乙醇性脂肪肝过量摄入乙醇往往会引起脂肪肝的变化。乙醇中毒者脂肪肝的发生率可达70%-80%。此种脂肪肝主要是肝内中性脂肪的增加造成的。中性脂肪的来源包括经口摄取的脂肪、外周组织中的脂肪和肝内所合成的脂肪。
乙醇性脂肪肝形成机制包括:①急性乙醇中毒时脂肪肝动员的增加,一次大量摄入乙醇,通过儿茶酚胺的作用引起储存脂肪的动员,同时伴有高脂血症的发生;②慢性摄取乙醇时乙醇代谢亢进,NADH/NAD+比值上升,使磷酸二羟丙酮向α-磷酸甘油的转化增加,有利于肝内甘油三酯的大量合成;③由于NADH的增加,NAD+的减少,导致三羧酸循环及脂肪酸的氧化被抑制,肝中脂肪酸的氧化降低;④大量摄取乙醇时脂蛋白的合成及分泌减少。
乙醇性脂肪肝是良性的可逆性的病态,它可因戒酒而消退,可以认为乙醇性脂肪肝是由乙醇代谢亢进引起的代谢紊乱造成的后果。
⒉乙醇性肝炎乙醇性肝炎在病理组织学上以肝细胞坏死为主要变化,伴有透明小体形成。关于此种肝细胞坏死的机制可能如下述:①乙醇性肝损伤时的蛋白贮留和蛋白的分泌障碍:乙醇性肝损伤时不仅肝内脂肪量增加,且肝细胞液中有蛋白(白蛋白、运铁蛋白等)贮留;②线粒体与内质网的损伤:乙醇及乙醛均作用于线粒体,造成线粒体损伤,后者与肝细胞坏死有关,长期摄入乙醇后MEOS这一代谢体系由平时占总代谢量的1/4升至1/2,所以内质网内乙醛的产生增加,也造成内质网的损伤;③乙醇在微粒体氧化时,氧自由基使脂质过氧化,导致肝中还原型谷胱甘肽的减少和过氧化脂质的增加;④乙醇引起的代谢亢进状态造成耗氧量的增加。⑤乙醇性肝损伤时,会出现IgA增多、白细胞粘着能力降低等免疫功能异常。
⒊胎儿性乙醇综合征胎儿性乙醇综合征是由于孕妇饮酒造成的胎儿异常,这是一种包含智能障碍的中枢神经系统的功能障碍,由出生前开始的发育障碍以及特有容貌和畸形为特征的综合征。FAS的动物模型表明其胎儿体重、大脑重量均较对照组为低,C-亮氨酸向脑核蛋白的掺入减少,脑内总RNA及tRNA减少。这表明FAS动物脑内蛋白质合成的抑制可解释人类FAS中智能发育延迟的现象。长期大量饮酒不仅危害人类的健康,还影响下一代的发育成长。
二、肝硬化的生化
肝硬化与病毒性肝炎、慢性酒精中毒、中毒性肝损伤有密切关系,在肝硬化的基础上有发生肝癌可能性。
肝硬化时可发生糖代谢异常,如肝糖原的减少,线粒体代谢障碍,使得乳酸、丙酮酸及α-酮戊二酸增多。脂类代谢中胆固醇酯的合成障碍,肝硬化晚期胆固醇的合成亦发生障碍。在蛋白质代谢方面,白蛋白、纤维蛋白原等的合成减少,氨基酸代谢及尿素合成异常,导致尿中出现氨基酸和血氨升高。
此外,肝硬化时肝的生物转化及对激素灭活的功能亦发生障碍。在肝硬化形成过程中,纤维组织形成是重要的形态变化。在缺氧和炎症刺激下,胶原纤维的合成增强。当发生肝硬化时,肝内的酸性粘多糖增加,特别是含有半乳糖胺的硫酸软膏素B显著增加,并与胶原纤维化有关,肝纤维化时增加的胶原以Ⅰ型及Ⅱ型胶原为主。
近十年来关于肝纤维化的研究有较大进展,发现许多与肝纤维化有关的因子,就细胞成分而言,枯否细胞(Kupffer cell,KC)、储脂细胞(fat-storing cell,FSC)在肝纤维化形成过程中都起着非常重要的作用,成纤维细胞与肝门静脉区的结缔组织形成有关。此外,胶原(特别是Ⅰ型、Ⅲ型及Ⅳ型)、弹性蛋白以及细胞外基质(其中所含的结合蛋白质及粘多糖)和某些相关的酶(如金属蛋白酶、胶原酶、透明质酸酶),还有一些肝纤维化的活化因子(如白细胞介素Ⅰ、肿瘤坏死因子、转化生长因子-βTGF-β、干扰素、前列腺素2等)等都与肝纤维化有关。
肝纤维化时枯否细胞可分泌多种细胞因子及胶原酶等生物活性物质,并由此对肝细胞外基质的合成与降解起调控作用,从而在肝纤维化的发生发展中起着重要作用。曾认为储脂细胞的活化是肝纤维化的关键,储脂细胞产生胶原,细胞外基质中的糖蛋白-板层素(LN)及纤维连接蛋白(FN)等的能力使得它在肝损伤后组织修复及纤维化的发生中起重要作用。探讨这几种细胞对细胞外基质形成的影响,阐明胶原合成的调节机制,有助于阐明肝纤维化、肝硬化的生化机制。
三、肝昏迷的生化机制
肝昏迷又称肝性脑病,是由严重的肝病所致的中枢神经系统功能紊乱,出现一系列精神症状直至进入昏迷。肝昏迷是肝功能不全的重危合并症。引起肝昏迷的代谢因素极为复杂。表10-16列举肝昏迷时血液成分的变化,可以看出肝昏迷时的代谢异常是多方面的。
目前认为氨基酸的代谢障碍可能是肝昏迷发生的生物化学基础。下面从氨中毒、假神经递质及氨基酸不平衡三方面来叙述肝昏迷的生物化学机制。
表10-16 肝昏迷时血液成分变化所反映的代谢异常
| 代谢指标 | 变化 |
| ⒈ 含氮代谢物 | |
| ⑴血氨 | 增高 |
| ⑵血液酚,吲哚 | 增高 |
| ⑶血浆氨基酸 | |
| 芳香氨基酸 | 增加 |
| 支链氨基酸 | 减少 |
| ⑷血清胺类 | 增加 |
| ⑸核苷类 | 减少 |
| ⑹未结合胆红素 | 增加 |
| ⑺粪卟啉 | 增加 |
| ⒉糖代谢 | |
| ⑴血糖 | 减少 |
| ⑵丙酮酸,乳酸 | 增加 |
| ⑶α-酮戊二酸 | 增加 |
| ⒊脂代谢 | |
| ⑴血清游离脂肪酸 | 增加 |
| ⑵血清短链脂肪酸 | 增加 |
| ⒋电解质及酸碱平衡 | |
| ⑴血清Na+ | 减低 |
| ⑵血清K+ | 减低 |
| ⑶血清pH | 升高或减低 |
㈠氨中毒与肝昏迷
40多年前即发现肝昏迷与氨代谢有密切关系,其依据是:①多数肝昏迷病人血氨增高,病情好转时血氨降低;②慢性肝病病人摄入高蛋白膳食或含铵药物,常诱发肝昏迷;③肝昏迷病人脑电图变化与血氨平行;④对门腔静脉吻合犬给以高蛋白可引起血氨升高及昏迷症状;⑤给猴注射醋酸铵,其血氨达200-400μg/dl时,显示与人的肝昏迷相似的症状;⑥上述病人及实验动物经过降血氨疗法病情常可好转。上述事实都表明氨中毒与肝昏迷有着密切关系。
关于氨中毒的机制,据认为是由于肝功能不全情况下,血氨的来源增多或去路减少,引起血氨升高,脑组织对氨毒性极为敏感,因而出现脑功能障碍而导致昏迷。正常血氨来自肠菌产氨(每日约4g)、肾泌氨、肌肉组织产氨等,而解除氨毒性的机制主要靠肝内的尿素合成。由于肝严重病变导致肝功能不全,清除氨的能力大为降低,加之门腔静脉短路,使由肠管回血液的氨不经肝解毒而直接进入体循环,造成高氨血症与肝昏迷。饮食蛋白过多、消化道出血、摄入铵盐、放腹水以及应用利尿剂等均可引起血氨的升高或氨毒性增加,从而能诱发肝昏迷。
氨对脑组织的毒性作用在于氨主要是干扰了脑的能量代谢,使高能磷酸化合物(ATP等)浓度降低。氨对脑细胞代谢的干扰有下述几方面:①氨能抑制丙酮酸脱氢酶的活性,影响乙酰辅酶A的生成,既干扰了三羧酸循环的起始步骤,又影响了神经递质乙酰胆碱的生成;②氨中毒时,脑内以形成谷氨酰胺的方式解毒,从而消耗了较多的NADH(α-酮戊二酸经还原性氨基化而生成谷氨酸),影响线粒体氧化磷酸化的正常进行,妨碍ATP生成;③大量氨与α-酮戊二酸结合生成谷氨酸,可使三羧酸循环中的α-酮戊二酸耗竭,妨碍了供能物质在脑细胞中能量的释放与转换。由于α-酮戊二酸及草酰乙酸难于通过血脑屏障,脑内转氨酶活性低,难于使α-酮戊二酸等得到补充,因此氨中毒使脑细胞三羧酸循环发生障碍,ATP的生成减少;④氨和谷氨酸合成谷氨酰胺时增加ATP消耗;⑤氨能激活神经细胞膜上的Na+、K+-ATP酶,并和K+有竞争作用,影响离子分布和神经传导的正常进行。
应当指出,氨中毒现象并不能解释所有肝昏迷的发生,有些病例血氨并不高,降血氨疗法亦不一定有效,尚须探讨其他机制。
㈡假神经递质学说
在肠管内,一部分氨基酸经肠菌的氨基酸脱羧酶作用而形成胺类,如苯丙氨酸及酪氨酸脱羧形成苯乙胺及酪胺,正常情况下可被肝内单胺氧化酶分解而清除。肝功能不全时,由于肝内单胺氧化酶活性降低或门体侧支循环的形成,于是芳香胺类直接经体循环入脑,经脑内非特异羟化酶作用,于是苯乙胺羟化而生成苯乙醇胺,酪胺经羟化而生成?J胺(β-羟酪胺)由于苯乙醇胺及?J胺与儿茶酚胺递质(多巴胺、去甲肾上腺素)结构相似,又不能正常地传递冲动,故称假神经递质。
假神经递质被释放后引起神经系统某些部位(如脑干网状结构上行激动系统)功能发生障碍,使大脑发生深度抑制而昏迷。黑质、纹状体通路中的多巴胺被假递质取代后,使乙酰胆碱的作用占优势,因而出现扑翼样振颤,当然,假递质学说也不能解释全部肝昏迷的发生机制(图10-3)。
㈢氨基酸不平衡与肝昏迷
近20年来关于肝昏迷时氨基酸代谢异常的资料表明:在严重肝功能损伤和有门腔静脉短路的条件下,由于种种原因引起体内氨基酸代谢异常。最突出的表现是血中支链氨基酸(缬氨酸、亮氨酸、异亮氨酸)浓度明显降低,芳香族氨基酸(苯丙氨酸、酪氨酸、色氨酸)明显增高,从而引起脑功能障碍。
由于芳香族氨基酸主要在肝内分解,当肝功能不全时,芳香族氨基酸在肝内代谢发生障碍,因而在血中的浓度增高;支链氨基酸主要在肌肉组织中代谢,由于肝功能不全时胰岛素的灭活发生障碍,在高水平的胰岛素的作用下,支链氨基酸大量进入肌肉组织被分解,因此血浆中的支链氨基酸的浓度降低。
芳香族氨基酸进入脑组织,可引起假神经递质的产生增多,色氨酸可使5-羟色胺的生成增
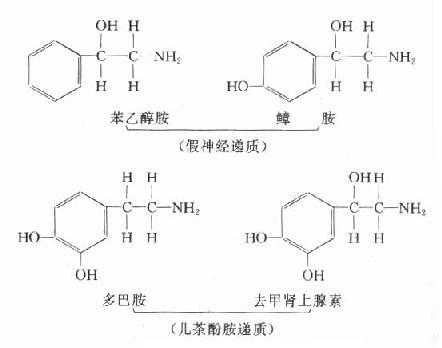
图10-3 假递质学说
多,后者为抑制性神经递质,使中枢进入抑制状态。而支链氨基酸的降低使得血脑屏障上芳香族氨基酸占了优势,缺少了竞争载体的对象,于是大量进入脑内,上述氨基酸代谢的不平衡引起严重的后果。上述三种学说可能解释不同情况下肝昏迷发病机制的主要方面。
肝昏迷(肝功能不全)的检验所见可有:①血清胆红素可呈显著的高值(能达40mg/dl),说明有胆汁排泄障碍;②血清白蛋白减低,表明其蛋白合成能力低;③低胆固醇血症,亦是合成能力降低所致;④AST及ALP由高值转为低值,见于大量肝细胞坏死的情况;⑤BUN呈低值,表示肝合成尿素功能低下;⑥血糖降低,由于肝糖原储备减少;⑦凝血酶原时间延长是由于肝中凝血酶原、Ⅶ因子、Ⅸ因子、Ⅹ因子的合成减低;⑧血浆纤维蛋白原呈低值,在肝内合成减低;⑨血氨增高是尿素合成降低所致;⑩血液pH增高,PCO2降低(呼吸性碱中毒)是因脑水肿引起的换气过度所致。
四、胆石症的生物化学
胆石症的主要组成成分有胆固醇、胆红素、钙及其他无机元素、胆汁酸、结石基质(硫酸化糖蛋白及糖蛋白)等。按胆石的主要成分及形成机制的不同,可将胆石分为胆固醇系结石、胆红素结石及其他胆石三大类,仅将前两大类胆石的形成机制略作介绍。
㈠胆固醇系结石的形成机制
胆固醇系胆石病人的胆汁可有下述异常:
⒈胆汁中胆固醇含量增高含有过饱和胆固醇的胆汁是胆固醇沉淀的先决条件。胆汁中胆固醇的需要量和胆汁酸盐及卵磷脂的含量保持一定的比例。如果胆固醇含量过高或胆汁酸盐及卵磷脂的含量降低,破坏了它们之间的正常比例,就形成了“致石性胆汁”。有报告指出:胆固醇系胆石病人肝内的HMGCoA还原酶(胆固醇合成的限速酶)活性增加,而7α-羟化酶(胆汁酸合成的限速酶)活性降低,即胆固醇的合成增加,胆固醇向胆汁酸的转化减少,这是形成胆固醇过饱和胆汁的代谢原因。
⒉胆汁中胆汁酸盐的减少胆固醇系胆石病人的总胆汁酸代谢池甚小,仅为正常人的一半。这一方面是由于其胆固醇代谢的失调,另一方面可能是由于胆囊对胆汁酸的重吸收增加及肠肝循环障碍所致。此外,必需脂肪酸的缺乏或代谢障碍可导致胆汁酸生成障碍。
⒊胆汁中胆汁酸组成的改变正常人胆汁中胆酸、鹅脱氧胆酸及脱氧胆酸三者的比例为1.3:1.0:0.6,而胆固醇结石病人的胆汁中鹅脱氧胆酸的比例则明显减低。
⒋胆汁中的磷脂降低胆固醇结石病人胆汁中的磷脂只为正常胆汁中的1/3。磷脂/胆固醇比值在正常胆汁为6.6,而在胆固醇结石病人的胆汁则为2.3。胆汁中的磷脂(90%是磷脂酰胆碱)是与胆汁酸盐、胆固醇共同形成混合微团的重要成分,它也与增加胆固醇在胆汁中的溶解有关。
⒌δ电位的降低 δ电位是混合微团吸附层与扩散层正、负离子分布均匀处两点间的电位差,电位差越大,则微团带电荷越多,稳定性越大。正常胆汁中甘氨胆汁酸与牛磺胆汁酸的比例约为3:1,胆固醇结石病人胆汁中牛磺胆汁酸减少,这个比例可达15:1,因而对胆汁酸的δ电位有影响。增加牛磺胆汁酸则可因其负电荷较强的-CH2SO3根而增加胆汁中的δ电位。
在肝生成异常胆汁后,在胆囊中形成肉眼可见的胆石,并逐步长大。在结石形成过程中,作为结石基质的糖蛋白类可能起着把胆固醇结晶及颗粒粘连在一起的网架作用。
㈡胆红素系胆石的形成机制
肝分泌的胆汁中的胆红素是结合型的葡萄糖醛酸胆红素。胆汁中由组织产生的β-葡萄糖苷酸酶活性降低(该酶最适pH5左右,胆汁pH为6.1-8.6),而且受该酶的抑制物葡萄糖二酸-1,4-内酯的抑制,所以正常胆汁中的葡萄糖醛酸胆红素不易被水解而处于良好的溶解状态。胆红素钙结石病人的胆汁中可出现下述异常:
⒈由于蛔虫钻进胆道,带入大肠杆菌,造成胆道感染,胆汁中出现细菌性β-葡萄糖苷酸酶(最适pH6.8-7.2,与胆汁pH一致)的活性高,超过了胆汁中该酶的抑制物β-葡萄糖二酸-1,4-内酯所能抑制的能力,因而可使葡萄糖醛酸胆红素大量被水解,生成游离胆红素而易于沉淀。
⒉胆红素系结石病人胆汁中葡萄糖二酸-1,4-内酯的含量降低在正常对照组胆汁中该物质的含量为200μg/ml,而胆红素钙结合病人胆汁中则仅含40μg/ml,这也是其胆红素易于沉淀的原因,因为β-葡萄糖苷酸酶抑制物的降低更有利于细菌性β-葡萄糖苷酸酶发挥作用。
⒊δ电位降低这是胆红素钙融合集结的条件。钙、钠、钾、镁等无机离子及胆道感染时出现的高分子有机物质,均可使胆盐微团的δ电位降低。
在胆红素结石形成过程中蛔虫残体、蛔虫卵及其他异物均构成结石的核心,细菌性β-葡萄糖苷酸酶催化葡萄糖醛酸胆红素的水解,游离胆红素与Ca+形成胆红素钙,胆红素钙又在前述无机离子的作用下,再加上结石基质(主要是硫酸化糖蛋白)的网架作用,而集结成胆红素钙结石。
五、肝癌的生化机制
㈠某些致癌物质与肝癌发生的关系
⒈乙酰氨基芴(AAF)AAF在肝内经生物转化后被活化成硫酸AAF,与蛋白质分子中的蛋氨酸残基及核酸分子中的鸟嘌呤碱基结合,引起生物高分子的结构与功能的异常。它与DNA结合,使细胞内调控蛋白的合成不足,细菌失去控制,但DNA复制不受影响,从而引起癌变。
⒉黄曲霉毒素 黄曲霉毒素B1被摄入体内后,在肝细菌微粒体混合功能氧化酶催化下转化成环氧化黄曲霉毒素B1,它可与DNA、RNA结合而致癌。
⒊二甲基氨基偶氨苯(DAB,奶油黄)可引起大鼠实验性肝癌。DAB在肝细胞混合功能氧化酶作用下N-脱羟化,再在硫酸转移酶作用下形成N-SO3-O-甲基氨基偶氮苯。后者再与核酸的鸟嘌呤碱基结合而引起癌变。
㈡肝癌时的代谢变化
⒈蛋白质及氨基酸代谢的变化癌组织中蛋白质合成旺盛,宿主其他组织蛋白质的分解增强。肝癌时,肝癌组织中与氨基酸分解代谢有关的酶(如色氨酸吡咯酶、酪氨酸转氨酶、苏氨酸脱水酶、苯丙氨酸转氨酸及组氨酸酶等)活性显著降低,表明癌组织中氨基酸分解代谢减弱,可使氨基酸重新用于蛋白质的合成,此现象可能与癌组织的生长有关。另外,与氨基酸转运有关的γ-谷氨酰转肽酶活性在肝癌组织中显著增高。
在肝癌组织中与尿素合成有关的肝组织特异酶-鸟氨酸氨甲酰基转移酶(OCT)、氨甲酰磷酸合成酶I(CPSI)、精氨酸酶等的活性降低;与核酸合成有关的天冬氨酸氨甲酰基转移酶(ATC),与细胞增殖及多巴胺合成有关的鸟氨酸脱羧酶(ODC)的活性则增高。在肝癌组织中支链氨基酸转氨酶的活性亦增高,这可能与供能有关。肝癌组织中白蛋白的合成降低。上述事实表明;在肝癌细胞中与增殖有关的酶活性增高,与肝细胞特异性功能有关的酶活性降低。肝癌组织还大量合成甲胎蛋白,呈现肿瘤组织的反分化特征。
⒉肝癌时糖代谢的变化肝癌组织中与糖代谢有关的酶活性呈下述变化:①糖异生关键酶(磷酸烯醇式丙酮酸羧激酶、果糖-1,6-二磷酸酶、葡萄糖-6-磷酸酶等)活性降低,癌瘤恶性程度越高,这些酶活性越低;②糖酵解酶系(已糖激酶、磷酸果糖激酶、丙酮酸激酶等)活性增高,癌瘤恶性程度越高,这些酶的活性越高;③同工酶谱的变化呈现胚胎化,能被调节的高Km型同工酶(已糖激酶Ⅰ-Ⅲ型等)的活性上升。肝癌组织中同工酶谱的变化可使ATP失去对糖酵解的调节作用,这可能是癌细胞失去巴斯德效应的原因之一。肝癌时糖代谢变化的主要特点是:糖的有氧氧化降低(正常肝有氧氧化占99%,酵解占1%,肝癌时酵解可占50%),糖酵解增加,糖异生减少,磷酸戊糖途径的代谢增强(表10-17)。
⒊肝癌时的脂类代谢在肝癌细胞中可发现磷脂的减少和甘油三酯的增加。在脂质组成中非脂肪酸增加,构成脂质的脂肪酸中出现C20:4的减少和C18:1的增加。在人的肝癌组织中能检查出非生理性的不饱和脂肪酸,其中最多见的是C20:39。此种不饱和脂肪酸只在血中AFP阳性的肝癌者中出现。
⒋其他据报道:大鼠原发性肝癌、移植性肝癌、癌前期等的肝细胞和正常肝细胞的Na+、K+-ATP酶活性的测定结果是:原发性肝癌和移植性肝癌细胞中该酶的活性都显著高于正常肝细胞,癌前期肝细胞的Na+、K+-ATP酶活性则高于正常肝细胞而低于癌细胞,提示了肝细胞癌变过程中钠泵活性的变化规律。肝癌细胞cAMP合成能力大为降低,肝癌组织中腺苷酸环化酶活性明显低于正常肝组织。
表10-17 正常肝与肝癌组织糖代谢有关酶活性的比较
| 比活性 | |||
| 代谢途径 | 酶 | ||
| 正 常 肝 | 迅速生长的肝癌组织 | ||
| 已糖激酶 | 100 | 500 | |
| 糖酵解 | 磷酸果糖激酶 | 100 | 229 |
| 丙酮酸激酶 | 100 | 449 | |
| 葡萄糖-6-磷酸酶 | 100 | <1 | |
| 糖异生 | 果糖-1,6-二磷酸酶 | 100 | <1 |
| 磷酸烯醇式丙酮酸羧激酶 | 100 | <1 | |
| 丙酮酸羧化酶 | 100 | <1 | |
| 磷酸戊糖通路 | 6-磷酸葡萄糖脱氢酶 | 100 | 751 |
| 糖酵解/糖异生 | 已糖激酶/葡萄糖-6-磷酸酶 | 100 | 8800 |
| 磷酸果糖激酶/果糖-1,6-二磷酸酶 | 100 | 6463 | |
㈢肝细胞癌变原理
关于细胞癌变的原理除了前述化学致癌物对于细胞的DNA的损伤之外,近10余年来随着癌基因与抑癌基因(抗癌基因)研究的进展,人们更加注重肝癌的分子生物学的研究,细胞内本来存在的原癌基因在物理、化学、生物性的致癌因素作用下被激活,通过点突变、基因易位、基因扩增等机制激活后而呈现过度表达,导致更多的癌基因产物(癌蛋白)的产生,造成细胞内基因表达调控的失常,最终导致细胞癌变。在这一癌变过程中不仅有原癌基因的变化,同时伴有抑癌基因的缺失或失活,并且癌变还是多阶段一步步地发生,这即多阶段癌变学说。所谓多阶段癌变学说即是说机体的癌变过程是长期多阶段地发生的,正常细胞在癌基因,抑癌基因等的异常的积累过程中,逐渐地离开了正常的细胞增殖控制机制的轨道,分阶段地向癌细胞转化,且恶性程度逐步增加。
近年来,关于肝炎(乙型及丙型)与肝癌的关系日益受到重视。乙型肝炎病毒(HBV)与肝癌的发生关系已基本阐明,HBV基因组中的X基因与肝癌有密切关系,用转基因小鼠实验证明此X基因可诱发肝癌的发生。X基因编码的X蛋白使肝细胞的DNA合成提高到发癌的准备状态,在多阶段癌变过程中起到促进作用。X蛋白具有促进DNA合成的作用,在肝细胞癌变过程中,X基因的高水平的持续性的表达是必要的,由于癌变过程中往往几种癌基因同时表达,因此人们都强调肝细胞癌的发生可能是多种癌基因协同作用的结果。如N-ras表达异常可导致跨膜信号传递的改变,进而启动了ets-2、C-myc、P53等核内表达产物异常,使细胞分裂增加,细胞处于活跃增殖状态。IGF-Ⅱ和fms表达增强可通过“自分泌”作用使细胞处于不断生长状态,多种因素综合作用最终导致细胞增殖失控而形成肝细胞癌,关于肝细胞癌的多阶段癌变学说如图10-4所示。
丙型肝炎病毒与肝癌的发生亦有密切的关系。丙型肝炎感染后20年左右导致肝硬化, 30年左右发展成肝癌,在日本肝癌有15%与乙型肝炎有关,有80%与丙型肝炎有关。本室用PCR技术检测肝细胞癌中乙型、丙型肝炎病毒核酸的结果,表明HBV-DNA阳性率为69.5%,HCV-RNA阳性率为30.4%,在我国丙型肝炎与肝细胞癌的关系亦正在日益受到重视。

图10-4 肝细胞癌的多阶段癌变学说
《临床生物化学》相关章节:
- ……
- 第十章 肝胆疾病的生物化学与实验诊断
- 第一节 概述
- 第二节 肝的生物转化功能
- 第三节 肝与胆汁酸代谢
- 第四节 胆红素代谢与黄疸
- 第五节 某些肝病的生化机制(当前内容)
- 第六节 肝细胞损伤时的肝功能试验
- 第十一章 肾功能不全的实验室生物化学诊断
- 第一节 概述
- 第二节 常见肾脏疾病的病理生物化学
- ……